分子轨道实际上就是一种近似处理,原子轨道线性组合形成分子轨道那就是进一步近似。核心在于Slater行列式和变分法。
单电子系统的薛定谔方程与分子轨道
对于单电子系统,做BO近似冻结原子核的运动,再扣除原子核之间的排斥能,于是就只剩下动能项和核与电子的吸引项,因此可以写出薛定谔方程(已使用原子单位):
其中 是电子与原子
的距离。该方程的解就是单电子系统的分子轨道。
变分法:用原子轨道的线性组合近似分子轨道
虽然像氢分子离子 可以精确求解其分子轨道,但是对于更大的系统我们却无能为力了。我们可以用一组原子轨道
的线性组合近似分子轨道
:
这种近似效果如何呢?变分原理告诉我们,任何一个符合边界条件的试探波函数的能量,总是不小于基态能量:
那么,调整系数 ,使得
的能量最小,就可以得到真实基态波函数的最佳近似。
这将化为久期行列式的求解(证明见附录):
其中,,
.
久期行列式可以解出 个
来,这实际上是
的
个极值,还可以证明,这些
分别对应第
个本征能量的上界:(证明见附录)
所以说用原子轨道的线性组合近似分子轨道是可行的。
实际上我们不必用准确的原子轨道作为基函数,对于原子轨道也可以进一步近似,这将衍生出基组的概念。
实例1:分子轨道理论处理氢分子离子
设 、
是两个氢原子的1s轨道,因此线性组合成分子轨道:
解久期行列式:
因为有 ,得到
最后一步用到了:
其中 、
、
皆与核间距
有关:
给大家画个图:
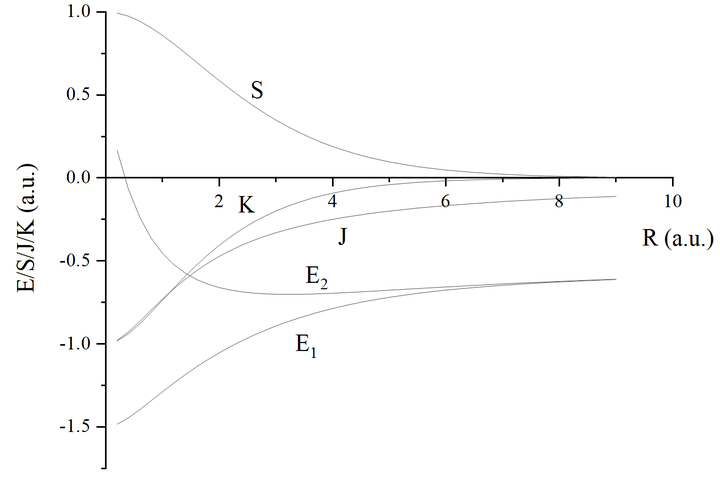
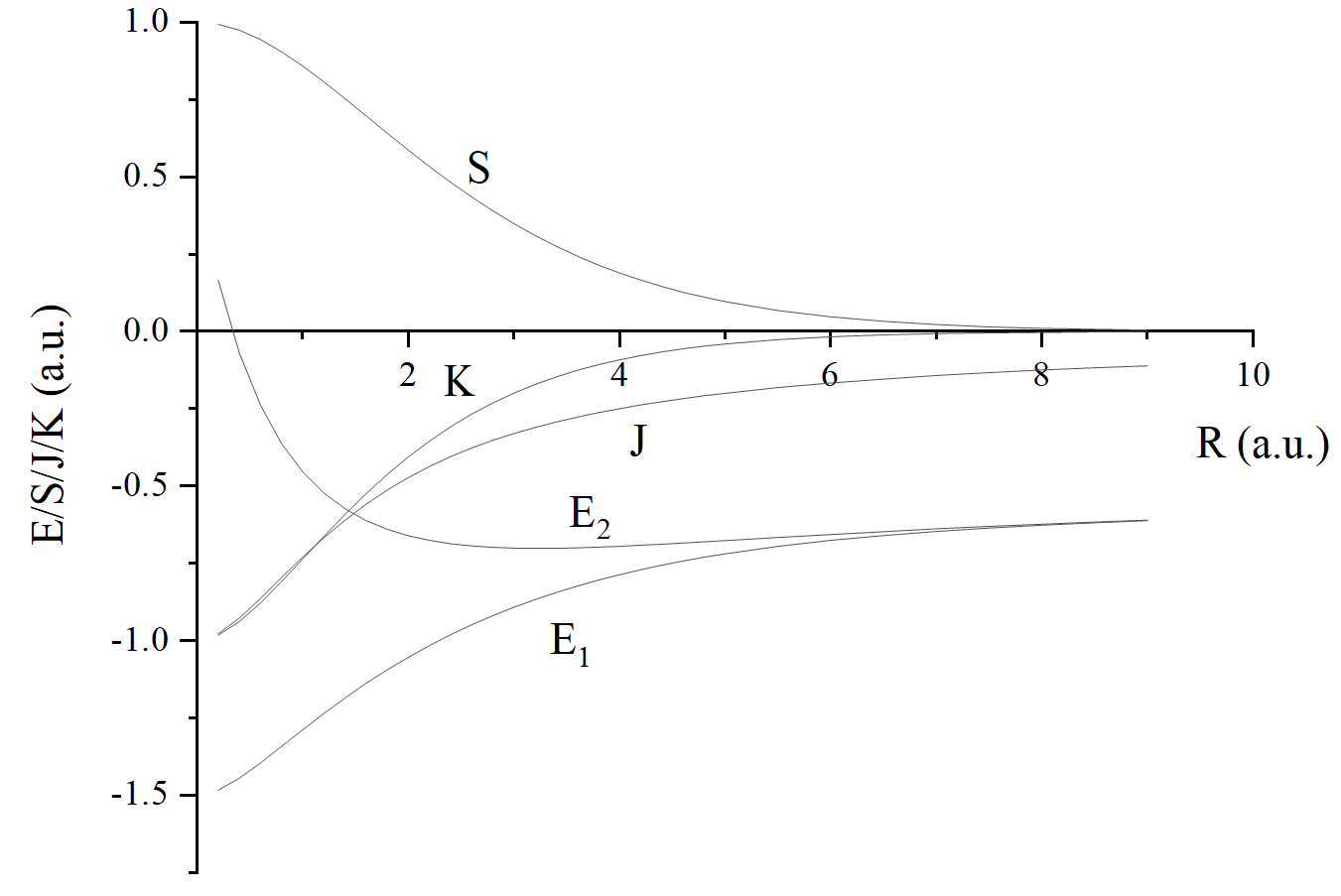
细心的读者会发现怎么反键轨道能量比原子轨道能量还低啊,对于氢分子离子,确实是这样。
进而得到波函数:
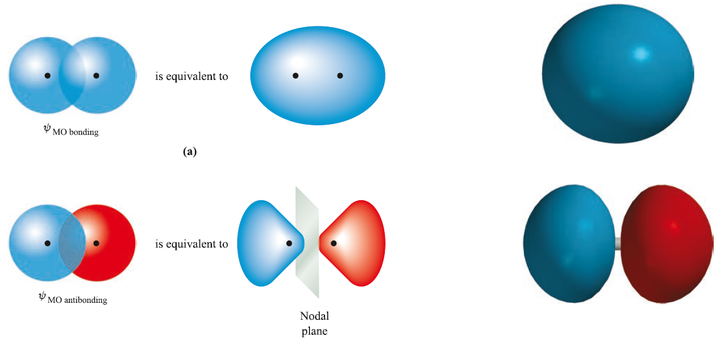
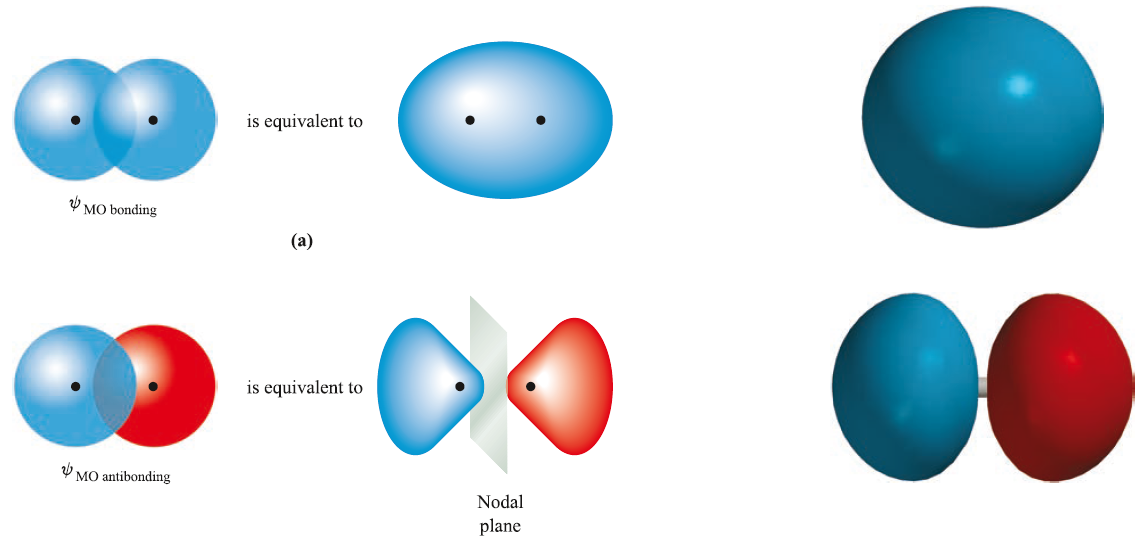
在此基础上加上原子核之间的排斥能 ,总能量曲线如下图所示:
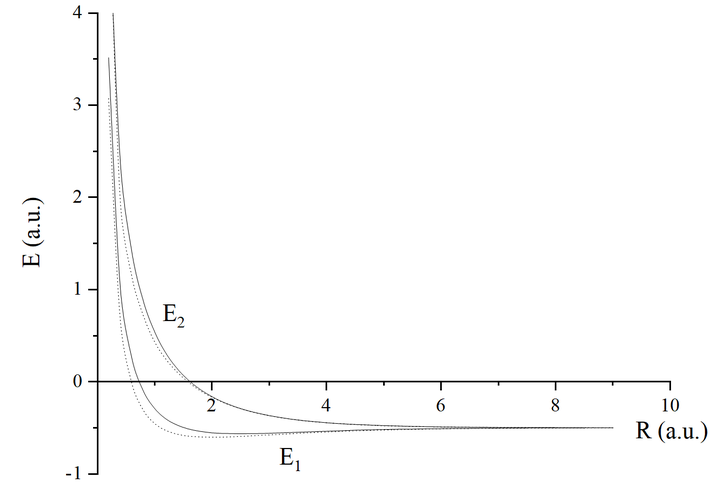
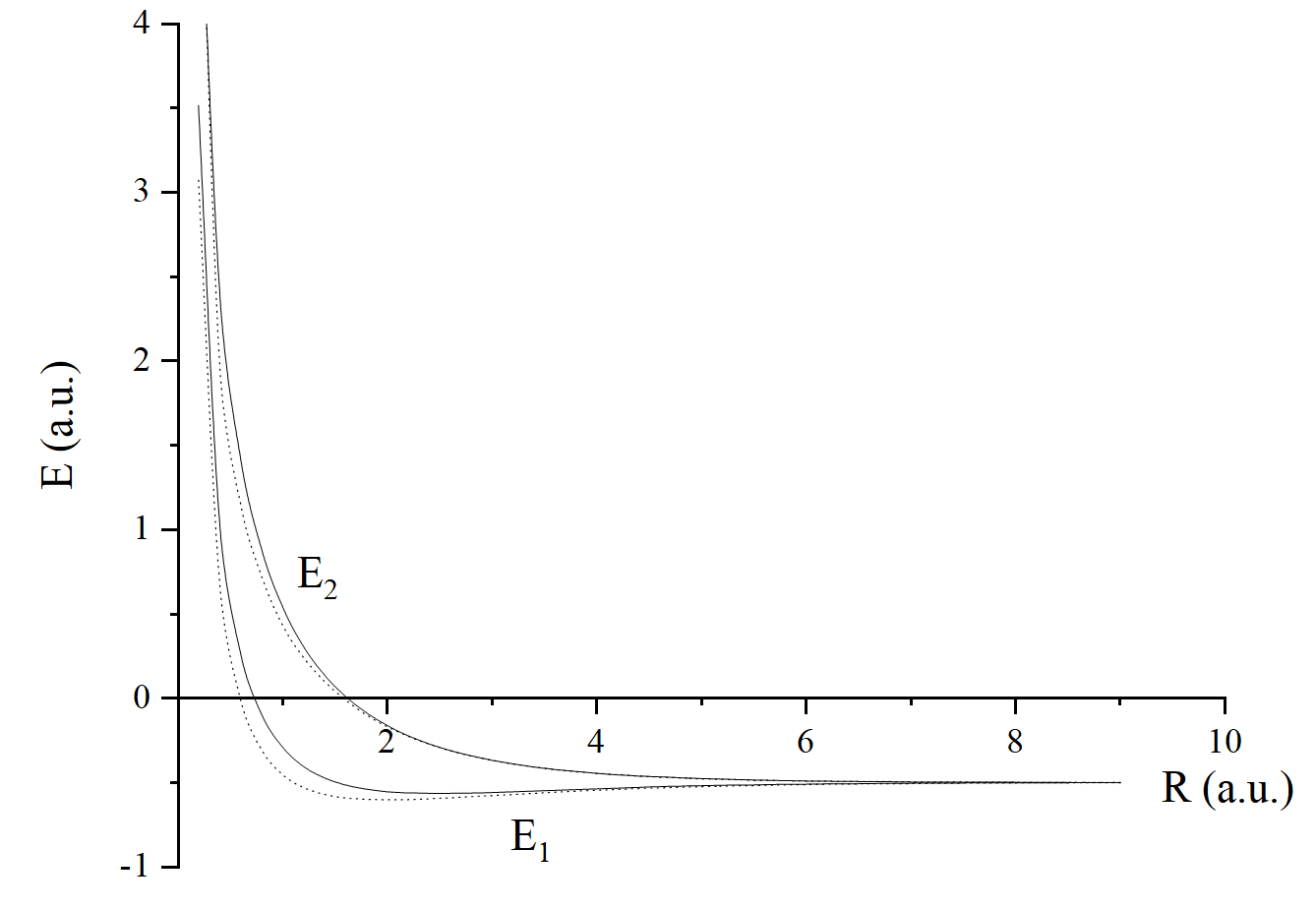
虽然变分法的结果(实线)与精确理论值(虚线)虽然定量上有所误差,但是给出了正确的定性结果。
Slater行列式
现在我们讨论多电子系统。
假设电子之间是没有相互作用的,那么薛定谔方程为:
显然,这是一个可分离变量的方程,分离变量后薛定谔方程可化为上面已经提到过的单电子薛定谔方程:
完整描述电子,光有分子轨道是不够的,还得乘上自旋波函数 :
于是总波函数变为
但是这个波函数这是不正确的,因为电子是费米子,具有交换反对称性:
上面的乘积并不满足这一条件。我们应该通过线性组合使得交换反对称性成立,我们自然而然可以想到,可以用行列式来表示波函数:
这就是Slater行列式,我们可以表记如下:
库仑积分与交换积分
对于有电子相互作用的系统,我们仍然使用Slater行列式去近似,但是如何寻找 呢,如何对无相互作用的单电子薛定谔方程进行修正呢?
在回答这个问题之前,我们先看一看有电子相互作用的Slater行列式的能量,B.O.近似去除原子核运动后的哈密顿算符为:
因此能量为
(式中对 的求和原本是
,但是因为
,所就重写成了
)
其中,
因此, 代表了无电子相互作用的轨道能量
,
代表了经典的库仑作用,称为库仑积分
,而
则只有在
和
自旋相同时才不为零,称为交换积分
。
交换积分体现出了自旋相关效应,自旋相同的电子具有相互远离的倾向,这导致了他们之间的库仑排斥减少,因此在库仑积分的基础上要再减去一个交换积分,这就是说,自旋相同的电子将导致能量降低,这将导出洪特规则。
最后我们将总能量重新写作:
实例2:分子轨道理论处理氢分子
显然,用两个原子轨道组成的两个分子轨道为
Slater行列式为:
氢分子的电子能量为:
其中 是氢分子离子的成键分子轨道的能量。注意,此处的
的下标与上一节推导中的下标含义不同,此处的下标是指分子轨道编号,而分子轨道与自旋波函数相乘得到的单电子波函数的编号。
接下来我们稍微探讨一下价键理论和分子轨道理论的关系,将波函数的空间部分拆开可得(以下推导省略归一化系数):
前两项 正是价键理论的结果,是共价项,而
则是两个电子全部偏于同一个原子的离子项。
可以预料到,这一波函数在近似两个氢原子的远距离行为时将会变得非常不准确,因为远距离时不应该具有离子项。
这里给大家看看一张HF/STO-3G计算结果和精确值的对比,体会一下这个现象:
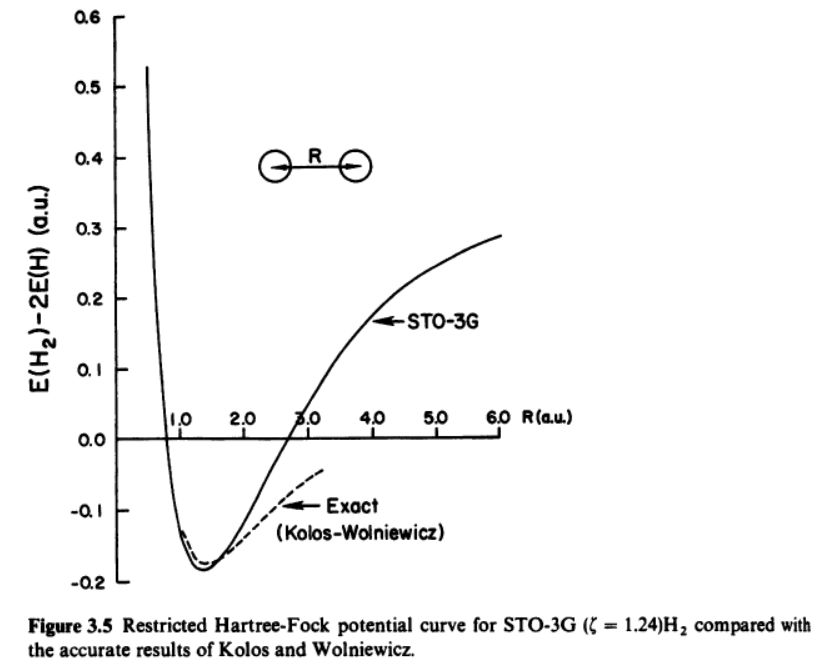
图中看似不满足变分原理是因为这是两个值相减的结果。
Fock方程:Slater行列式近似下分子轨道所满足的方程
由上分析可得,一个电子应当除了受到核的吸引、经典的库仑作用外还受到一种自旋相同电子间的交换作用。这样我们直接写出单电子波函数 的近似“薛定谔方程”,其实就是Fock方程(通过变分法推导Fock方程见附录):
其中交换算符 的作用是
简记为:
进一步简记为
将 去掉自旋波函数
,就得到了分子轨道(Hartree-Fock轨道)
。
分子轨道的能量为:
恭喜你,你发现了总能量并不等于分子轨道能量相加:
我们仍然可以用一组原子轨道 的线性组合近似Hartree-Fock轨道
:
同样地线性变分法的最佳近似解是通过求解如下方程得到的:
其中,,
.
我们将 个
和
个系数
组合起来写成如下两个矩阵:
就可以得到Hartree-Fock-Roothaan方程:
该方程可以通过迭代求得自洽解。
到此为止大家应该完全地理解分子轨道理论了,完结欢呼。
推荐书目
先学学结构化学,这个我不知道哪本最好。
然后学学量子力学,比如格里菲斯《量子力学概论》;顺便学学线性代数。
然后就可以看 Szabo & Ostlund, Modern Quantum Chemistry 了。
可以参考赖文《量子化学》、徐光宪等《物质结构》、徐光宪等《量子化学》。
另外还有一本 Albright et el., Orbital Interactions in Chemistry ,也不错。
附录
变分原理
设 是
的本征函数集,对于任意归一化的试探波函数
可以进行展开:
因此能量:
线性变分法
设试探波函数 为一组有限的基函数
的线性组合
为了方便讨论,假设系数都是实的。其能量为
能量取极值时,则能量对于变分参数的偏导数为 ,
(如果系数是复数,那么对于
和
的偏导数都要为零)。
先将分母乘到左边,然后求导,得到方程组:
整理为关于系数的方程:
注意到 因此上式可写为:
综合不同 得出的方程,可以写出:
为了使该方程组有非零解,需要系数矩阵的行列式(即久期行列式)为0,因此:
或者直接写出来就是:
这是一个 阶行列式,因此是
的
次方程,因此有
个解,对应地,就有
套归一化的线性系数
,就有
个分子轨道。
所以,有几个原子轨道参与组成分子轨道,就有几个分子轨道。
我们知道 ,但是在线性变分法中还有
,下面给出证明。
线性变分法中能量极值对应本征能量上界的证明
不妨设基函数 都是正交的,于是有
根据试探波函数能量极值的大小 ,记对应的试探波函数为
,记本征能量
对应的本质波函数为
,
现在令新的归一化试探波函数 ,并且满足
将 用本征波函数展开:
有
因此
而
故
变分法推导Fock方程
Slater行列式 的能量为:
我们要求其中的单电子波函数 都是正交的:
我们用拉格朗日乘数法求 的极值:
极值满足 。
现在对 添加微扰
,
二分之一系数之所以去掉了,是因为 ,
其中每一项的 为前一项的复共轭。
因此
由于 是任意的,故
也就是
接下来,通过酉变换 使
对角化即可得到:
完结撒花。
可能错误很多但是回答了题主其它问题的原回答
- 为什么可以用原子轨道线性组合表示分子轨道?
实际上是,因为我们无法解出更大体系的精确解,所以我们考虑先做BO近似和单电子近似,认为总波函数是由分子轨道组成的Slater行列式,然后用原子轨道的线性组合来近似分子轨道。
变分原理告诉我们,试探波函数的能量总是不小于基态能量,而线性变分法则进一步可以得出,解出的第n个能量极值不小于第n个本征态的能量。
所以n个原子轨道线性组合,可以求得n个能量极值,所以就得到n个分子轨道,这些分子轨道就分别用来作为真实本征态的近似。
更新:评论区大佬指出我上一句话“完全不对”,或许我应该说n个原子轨道线性组合按照变分法可得到n个近似分子轨道,作为准确分子轨道的近似?
- 以及轨道在坐标系中一定要取得四四方方么?比如对于甲烷,我们放好四个H之后歪着摆上C的原子轨道,那么得到的结果还是三重简并态么?
把原子轨道重新线性组合一下就可以得到摆正但仍然正交的轨道了。
- 以及确定形状的分子的轨道是一定的么?
综上,因为分子轨道是要求满足其为试探波函数的能量的极值,因此是一定的。
更新:仔细一想,氧分子中的π分子轨道可以“乱转”,这个是要看对称性。
- 推荐一些合适的书籍
先去找本《结构化学》看,然后学学线性代数后看看格里菲斯《量子力学概论》、Szabo & Ostlund, Modern Quantum Chemistry等等。
另外还有赖文《量子化学》、徐光宪等《量子化学》、Albright et el., Orbital Interactions in Chemistry